Definition
CADASIL (Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy) is a hereditary autosomal dominant disease affecting all the small cerebral arteries. It causes subcortical infarcts and damages the white matter (leukoencephalopathy) and it is due to various mutations of the Notch3 gene situated on chromosome 19.
Epidemiology
Initially described in Europe, the disease has now been observed in families with very different ethnic backgrounds, on all continents. At present, there are more than four hundred families in Europe. There has not yet been any real epidemiological study of CADASIL in France. The authors of a study carried out in the West of Scotland in 2002 listed 22 patients with CADASIL from seven families out of a population of 1,418,990. Considering the relatives of these patients, who risk being carriers of the mutated gene, the researchers estimated the prevalence to be 4.15/100,000 inhabitants. It is, though, likely that the frequency of the disease is as yet underestimated.
Clinical description
The initial clinical signs, which are observed in 20% to 30% of patients, are the onset of migraine with aura starting between the ages of 20 and 40. Cerebral infarcts (ischemic strokes) are observed in 70% to 80% of patients with onset usually around the age of 50. There are also cognitive disorders (difficulties with concentration and attention, memory loss), to a greater or lesser extent. These difficulties occur very early in the development of the disease but do not become significant until the 50 to 60 years age span. These cognitive difficulties may lead to a change in social life and, eventually, to almost constant dementia in the terminal phase of the illness, combined with difficulties in walking and balance. In 10% to 20% of cases, there are also psychiatric disorders and, in 5% to 10% of patients, there are epileptic seizures.
Migraines with aura (i.e. migraines accompanied by neurological signs) are reported by one in four patients The frequency of the migraines is extremely variable, ranging from twice a week to one every 3 or 4 years. Symptoms of the aura are, in order of frequency, visual, sensory, aphasic or motor. A visual aura manifests itself in various forms, most frequently as a scintillating scotoma and less often as blurring of vision or as a homonymous lateral hemianopsia. Speech disorders during attacks of migraine with aura can often be summarised as difficulties in expressing oneself, with reduced verbal fluency.
More than one-half of patients suffer migraine with atypical aura i.e. sudden-onset migraines with aura, « basilary » migraine or « hemiplegic » migraine. In a few cases, the migraines may be extremely severe such as those seen with familial hemiplegic migraine. They produce episodes of confusion, lack of vigilance, coma and hyperthermia (possibly lasting for several hours or several days).
Some 70% to 85% of patients report the occurrence of an ischemic event which can be a neurological deficiencyof sudden onset resolving in less than 24 hours (TLA transient ischaemic attack) or a permanent neurological deficiency. In most cases these signs indicate a minor stroke resulting in traditional signs (lacunar syndrome caused by the occlusion of a small artery: pure sensory deficit, pure motor deficit, or sensori-motor deficit of one side of the body or ataxic hermiparesis). These cerebral infarcts can occur in the absence of any of the usual vascular risk factors (arterial hypertension, diabetes, or hypercholesterolemia).
Mood disturbance are observed in one in five patients. They may be early (up to 10% of patients), sometimes inuagural and lead to an error or delay in diagnosis. Some patients describe symptoms of severe depression suggesting melancholia, alternating, in a few cases, with episodes of mania (this can lead to the presumptive diagnosis of bipolar disorder). Apathy (loss of motivation) is a frequent sign of the disease, depending on the location of the brain lesions. It is not always secondary to depression.
Cognitive disorders (difficulties with executive functions, attention and memory) are extremely frequent but of variable severity during the course of the illness. Alteration of the executive functions (planning, anticipation, adjustment, self-correction and mental flexibility) is the earliest symptom most frequently observed and it can be almost imperceptible for many years. Damage to the executive functions is frequently associated with attention and concentration disorders. Gradually, with age, the decline becomes more acute with the onset of apathy, often the most observable feature, and deficiencies in motor functions (tasks such as drawing or writing done using external resources), suggestive of diffuse cerebral damage. However, there is very rarely any severe aphasia (language difficulties), apraxia (difficulties with voluntary behaviour) or agnosia (difficulty with the recognition of objects, people or places with visual difficulties), all features frequently observed in Alzheimer's disease. Semantic memory (linked to knowledge) and recognition are often maintained. Cognitive decline commonly appears gradually, often in the absence of any ischaemic events. This development may therefore suggest a degenerative disease. Sometimes, the patient suddenly worsens, in stages.
Dementia (cognitive difficulties that affect the patient's everyday life and lead to a loss of independence) is observed in one-third of patients, especially after the age of 60. Its frequency increases with age and approximately 60% of patients over the age of 60 have dementia. It is often associated with other signs of the gravity of the disease e.g. difficulty with walking, urinary incontinence and, sometimes, a pseudo-bulbar palsy (difficult swallowing, spasmodic laughter or crying).
Despite the diffuse damage to small arteries in all organs, the clinical manifestations of the disease are only neurological and restricted to the brain.
Clinical course and prognosis
The typical progression of the disease begins with the onset of migraine with aura when the patient is in his 30's followed by transient or constituted ischaemic cerebral events a decade later and the gradual onset of cognitive difficulty, problems with balance and walking as the patient approaches the age of sixty. Loss of independence with motor and cognitive handicaps is frequent after the age of 60 (Figure 1)
This profile is not constant because of significant variability in the course of the disease, sometimes between several members of the same family (i.e. having the same genetic abnormality). In some cases, the disease can produce an early handicap at age of 40.Conversely, in other cases, the first signs of the disease may not appear until age of 70.
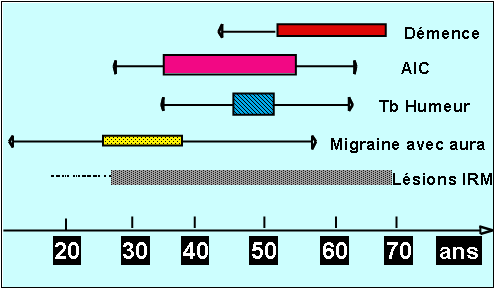
Figure1: A summary of the natural history of the disease
Diagnosis
Magnetic resonance imaging (MRI) is essential for the diagnosis of this disease. Abnormal MRI signals (abnormalities in the white matter of the brain) are sometimes detected before the onset of the first symptoms of disease. These abnormalities appear between the ages of 20 and 35 and can therefore remain inconsistent in this age group. On the other hand, after the age of 35, all the carriers of the mutated gene have MRI abnormalities suggestive of the disease, whether or not they have any symptoms. The total absence of MRI abnormalities after the age of 35 should cast doubt on the diagnosis.
Several types of abnormality may be observed (Figure 2).
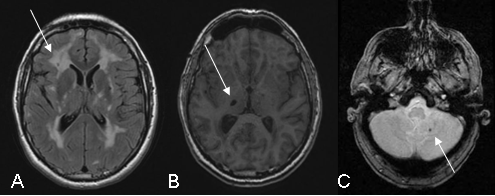
Figure 2: Illustration of the MRI abnormalities detected on the following sequences: FLAIR (A), T1 (B) and gradient echo (C)
White-matter hypersignals (A)is constant when major symptoms of the disease are present. They are observed on T2 weighted sequences which show extensive hyperintense areas within the white matter of the brain associated with more focal abnormalities within the deep gray nuclei, thalamus and brain stem. The extent of white matter hypersignals is variable and increases with age. In patients under the age of 40, signal anomalies are usually punctiforms or nodular and are symmetrically distributed. Gradually, as the disease develops, hypersignals become confluent and extend to the entire white matter. The presence of these signal anomalies in the anterior temporal lobes (more than 2 out of 3 patients) is very important from the diagnostic point of view because of their great specificity. They are usually not seen in cerebral arterial diseases caused by hypertension or diabetes.
Lacunar infarcts (B) are detected on T1 weighted images in the form of limited zones with hypointense signal. They are punctiform or wider depending on the cavity forming as a secondary feature after a minor infarct. These lesions are observed in approximately two out of every three patients with abnormalities in the white matter of the brain. They are present within the white matter, deep gray nuclei and brain stem. The total volume of these lesions correlates strongly with the clinical severity of the disease.
Microbleeds (C) are seen in one out of three patients, on average, using gradient echo sequences or (T2* sequences) since they are very sensitive to the accumulation of haemoglobin by-products in cerebral tissue. The bleeds do not usually produce any specific signs but their presence seems, in most cases, to be associated with greater damage to the vascular wall and greater severity of the disease.
Diagnosis
CADASIL is a hereditary familial disease. The mode of transmission is autosomal dominant (found with the same frequency in both male and female patients, 50% of children born to a person with the disease have the genetic abnormality) (Figure 3).
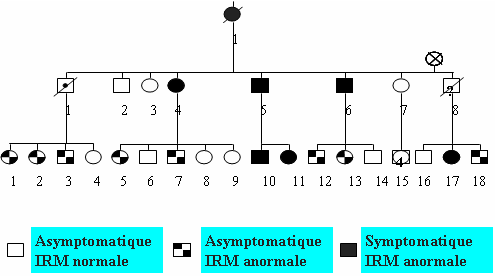
Figure 3: Family tree showing the autosomal dominant transmission and the results of the MRI scan.
The diagnosis should be discussed with patients who have symmetrical lesions in the white matter of the brain and a clinical history of migraine with aura, TIA or brain infarction, mood alternations or cognitive difficulties of unexplained origin.
It is essential to question patients and seek clinical histories in other members of the family suggestive of the disease. A history of multiple sclerosis (an incorrect diagnosis of multiple sclerosis is sometimes made for young patients after a first clinical event), cerebral vascular events or gradual-onset dementia with motor deficit in relatives should point to a family history of cerebral small vessels disease. However, the total absence of any family history should not lead to the diagnosis being discarded because of the possibility of a new mutation in the gene responsible, causing new, sporadic cases.
The presence on an MRI showing T2 or FLAIR hypersignal, with symmetrical distribution in the cerebral white matter, especially in the anterior temporal lobes increases the likelihood of diagnosis (CADASIL) because of the specific nature of these signs.
Testing for other causes of damage to the small cerebral arteries (standard blood test, search for an inflammatory syndrome, search for vascular risk factors with tests for hypercholesterolaemia, homocysteinaemia or fasting glucose, or ultrasound investigations of cervical and intracranial arteries) is usually negative.
If there is a strong suspicion of the diagnosis, a conventional angiography should be avoided because of the risk of severe neurological symptoms (severe headache, migraine with marked aura) which can, in some cases, be serious. This examination is usually normal, although it may sometimes show narrowing of the small arteries. An MRI scan is preferable if seeking to investigate the state of the medium and large arteries.
 To confirm the diagnosis, genetic testing must always be carried out. The gene involved is the Notch3 gene, situated on the short arm of chromosome 19. It consists of 33 exons including 23 exons (2 to 24) which encode for EFG-like motifs with six cysteine residues. To date, all the mutations responsible for the disease have been located within these exons (exons 2 to 24). The mutations are highly stereotypical and all of them lead to the addition or loss of one cysteine in one of the EGF-like motifs. The presence of a mutation of this type confirms diagnosis of the disease beyond all doubt. Within the French population, the mutation lies within exons 3 or 4 of the Notch3 gene in 70% of cases while in 90% to 95% of cases, the mutation is located in one of the following 12 exons: 2, 3, 4, 5, 6, 7, 8, 11, 12, 18, 19 or 20. In the absence of any known mutation in the patient's family, exons 3 and 4 (70% sensitivity) are tested first, followed by exons 2, 5, 6, 7, 8, 11, 12, 18, 19 or 20 (95% sensitivity). If there are very strong pointers to the diagnosis (hence the importance of sending the clinical data and MRI scan) and if the previous analysis has been negative, screening can be extended to the last mutated exons in the gene in a very small number of CADASIL patients. The sensitivity of the screening of 23 exons encoding for the EGF areas in the Notch3 gene is estimated to be close to 100 %.
To confirm the diagnosis, genetic testing must always be carried out. The gene involved is the Notch3 gene, situated on the short arm of chromosome 19. It consists of 33 exons including 23 exons (2 to 24) which encode for EFG-like motifs with six cysteine residues. To date, all the mutations responsible for the disease have been located within these exons (exons 2 to 24). The mutations are highly stereotypical and all of them lead to the addition or loss of one cysteine in one of the EGF-like motifs. The presence of a mutation of this type confirms diagnosis of the disease beyond all doubt. Within the French population, the mutation lies within exons 3 or 4 of the Notch3 gene in 70% of cases while in 90% to 95% of cases, the mutation is located in one of the following 12 exons: 2, 3, 4, 5, 6, 7, 8, 11, 12, 18, 19 or 20. In the absence of any known mutation in the patient's family, exons 3 and 4 (70% sensitivity) are tested first, followed by exons 2, 5, 6, 7, 8, 11, 12, 18, 19 or 20 (95% sensitivity). If there are very strong pointers to the diagnosis (hence the importance of sending the clinical data and MRI scan) and if the previous analysis has been negative, screening can be extended to the last mutated exons in the gene in a very small number of CADASIL patients. The sensitivity of the screening of 23 exons encoding for the EGF areas in the Notch3 gene is estimated to be close to 100 %.
The diagnosis can rarely be made by a skin biopsy (punch biopsy) which shows the status of small vessels. There are two possible approaches - a study of the vessels under an electron microscope showing the accumulation that is characteristic of the disease within the wall of small vessels, known as GOM (granula osmiophilic material), or a study using an anti-Notch3 antibody which, under the microscope, highlights the accumulation of Notch3 protein within the vascular wall. Both of these methods are highly sensitive but technically fairly difficult to use. At present, these tests are carried out less and less frequently because molecular testing has become easier.
Genetic diagnosis is possible before symptoms of the disease appear, in the other members of an affected family. However, genetic testing is only carried out on healthy subjects with no clinical signs of the disease who have not had any previous test within the setting of a specialist multidisciplinary consultation. After a neurological assessment (neurologist), a psychological evaluation (interview with a psychologist) and a genetic consultation (geneticist), the patient's request is assessed jointly by all the practitioners and a cooling-off period of several weeks is suggested before any blood test. The patient may request not to be informed of the results of the test throughout the procedure, until the final results are ready. Clinical and psychological follow-up are always proposed once the results have been given.
No genetic testing is currently carried out on minors who are symptom-free.
Etiology/pathophysiology
The symptoms of the disease are mainly produced by the lesions occurring within the brain as the disease progresses. The lesions observed in the white matter correspond to demyelination (loss of myeline sheaths which are manufactured by the oligodendrocytes in the white matter) and to a loss of axons in the brain's neurons. These lesions are associated with minor infarcts occurring mainly deep inside the brain as a result of an interruption in the blood flow to an area supplied by a small artery. The infarcts can leave a small cavity or hole known as a « lacune ». Traces of tiny haemorrhages may also be visible in one-third of patients. The latest cerebral imaging studies show that it is mainly the accumulation of minor infarcts in the brain that explains the severity of the disease during CADASIL.
The lesions in the white matter and the deep infarcts are due to a reduction in cerebral perfusion. A decrease in blood flow in the brain was observed within the white matter and sometimes, in a more diffuse manner, within patients' brains. Permanent reduction in blood supply (and, therefore, in oxygen provided by the red blood cells) would appear to become more severe as the disease progresses and this explains the gradual accumulation of cerebral lesions and the increasing acuteness of symptoms.
CADASIL is a disease affecting mainly the walls of the small arteries (arterioles) in the brain and other organs. In many cases, the artery walls thicken; in some, they become fibrous. The smooth muscle cells in the central layer of the vessel wall (media) are abnormal or are gradually disappearing. Around them, there is a granular substance called GOM (granular osmiophilic material) which is typical of the disease and visible under an electron microscope. The exact origin of the GOM deposits is currently unknown. Recent work has shown that part of the Notch3 gene, which is a receptor on the surface of the membranes of smooth muscle cells, builds up near the GOM in the vessel walls. Recent research in human subjects and mice with the genetic abnormality showed that the wall of the small arteries did not contract or dilate normally. It may be that the narrowing of certain vessels, in addition to this abnormal reaction, produces the loss of perfusion observed in CADASIL patients.
We do not yet know why mutations in the Notch3 gene, which lead to an abnormality in the Notch3 receptor of the smooth muscle cell in the blood vessel, also lead to a build-up of protein, the appearance of GOM and the degeneration of smooth muscle cells in the vessel wall. The important part played by the Notch3 gene in the development of small arteries has, however, been clearly demonstrated.
Treatment
No specific preventive treatment for this disease is known to date in CADASIL patients. Because of the occurrence of cerebral infarcts, aspirin is traditionally used as secondary prevention but the benefit of this treatment when the disease is already present has not been demonstrated. The possible occurrence of intracranial haemorrhages, although rare, suggests that the use of anticoagulants would, on the other hand, be risky.
For migraine with aura, vasoconstrictors are not recommended because of the theoretical risk of a reduction in cerebral blood flow in patients in a precarious haemodynamic condition with decreased cerebral blood flow. NSAIDs and analgesics are therefore recommended as first-line treatment of migraine.
The usefulness of acetylcholinesterase inhibitors was recently assessed as a means of helping patients with cognitive difficulties. This study found no significant treatment effect of donepezil on cognition as assessed by the primary efficacy measure but improvements were noted on several measures of executive function.
All the hypotensive treatments (neuroleptics, anti-hypertensives) must be used with care because of the possible risk of a decrease in cerebral blood flow in patients with reduced cerebral perfusion.
On the other hand, physiotherapy is essential and must be widely prescribed when motor signs and difficulties with walking and balance are present, especially after a stroke. Speech therapy is prescribed to improve communication and language abilities when necessary.
Psychological support is crucial at every stage of the disease, both for the patient and for the family and carers. It should include ways of dealing with the psychological consequences resulting from neurological deficiency, an assessment of psychological disorders directly linked to the disease, ways of dealing with the consequences of the handicap within the family unit and psychological counseling because of the familial and hereditary nature of the disease.
Research
Current research work covers two areas: 1) clinically, there is a need to define all the clinical and MRI parameters required to set up therapeutic testing in the future for a rare disease that develops slowly, over several decades, and to gain greater insight into prognostic factors and factors that might explain the variable degrees of severity of this disease, 2) there is a need for research into the molecular mechanisms that lead from the genetic abnormality in the Notch3 gene to the lesions observed in the walls of the blood vessels. This is being done using animal models of the disease.
1) clinically, there is a need to define all the clinical and MRI parameters required to set up therapeutic testing in the future for a rare disease that develops slowly, over several decades, and to gain greater insight into prognostic factors and factors that might explain the variable degrees of severity of this disease, 2) there is a need for research into the molecular mechanisms that lead from the genetic abnormality in the Notch3 gene to the lesions observed in the walls of the blood vessels. This is being done using animal models of the disease.